Research in the Lab
Our lab primarily uses snakes as model systems to study diverse questions about genome biology, functional innovation, natural selection, and speciation, although through collaborations we regularly work on other diverse systems ranging from schistosome parasites to venomous mammals to COVID. Research in the lab revolves around both the generation and analysis of diverse ‘omic’-scale datasets, including genomic, transcriptomic, phylogenomic, epigenomic, single-cell data, and other functional genomic data (e.g., ChIPseq, ATACseq, and Hi-C contact data). Our research is motivated by broad biological questions and driven by generation and analysis of empirical data, and these motivations often lead us to develop new theory and new approaches that have broad relevance beyond our own research. Below we outline major research projects driven by broad questions in the laboratory:
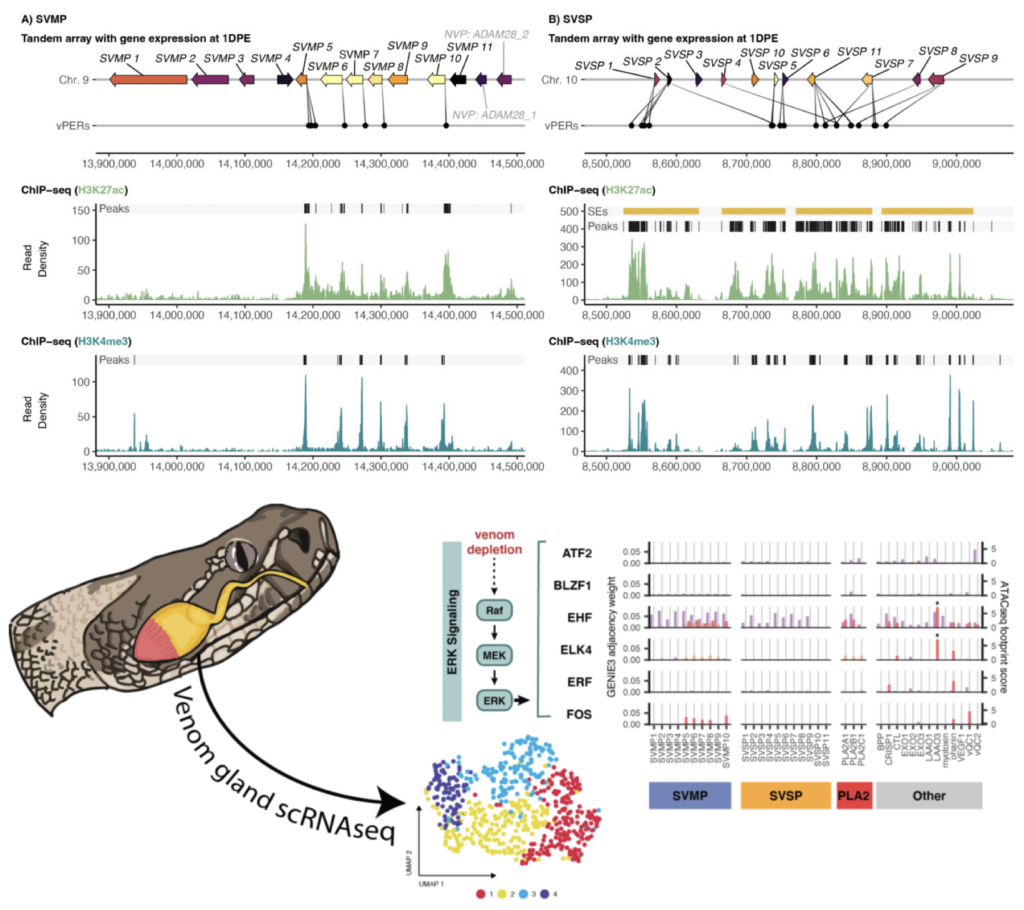
Using snake venom systems to understand how new gene regulatory networks arise and evolve
How are regulatory networks re-wired to accomplish novel functions and how does evolution shape the complex structure and function of gene regulation, and how does this manifest at cellular levels? A rapidly expanding area of research in the lab is using snake venom glands and venom expression as a model to understand how novel phenotypes arise through the evolution of new gene regulatory networks. This project is exciting because it unites our interests in population genomics and functional genomics, and has driven us to incorporate a diversity of new functional genomics approaches (Hi-C, ChIPseq, ATACseq), along with RNAseq and proteomics and single-cell approaches, to develop extremely detailed models of venom gene regulation. The overarching goal of this research program is to advance the ability to understand and predict how new gene regulatory networks arise and how variation in these networks shape complex physiological traits, using snake venom as a model. New methods developed will integrate predictive approaches and diverse functional genomic data to test mechanistic hypotheses and predict the roles of regulatory elements, trans-acting factors and other features that govern venom composition, and to test how cellular and evolutionary heterogeneity together shape organism-level phenotypes. Key products of this work include novel approaches that leverage both single-cell variation and evolutionary variation to test hypotheses for the roles of gene regulatory components that will be broadly applicable to any eukaryotic system.
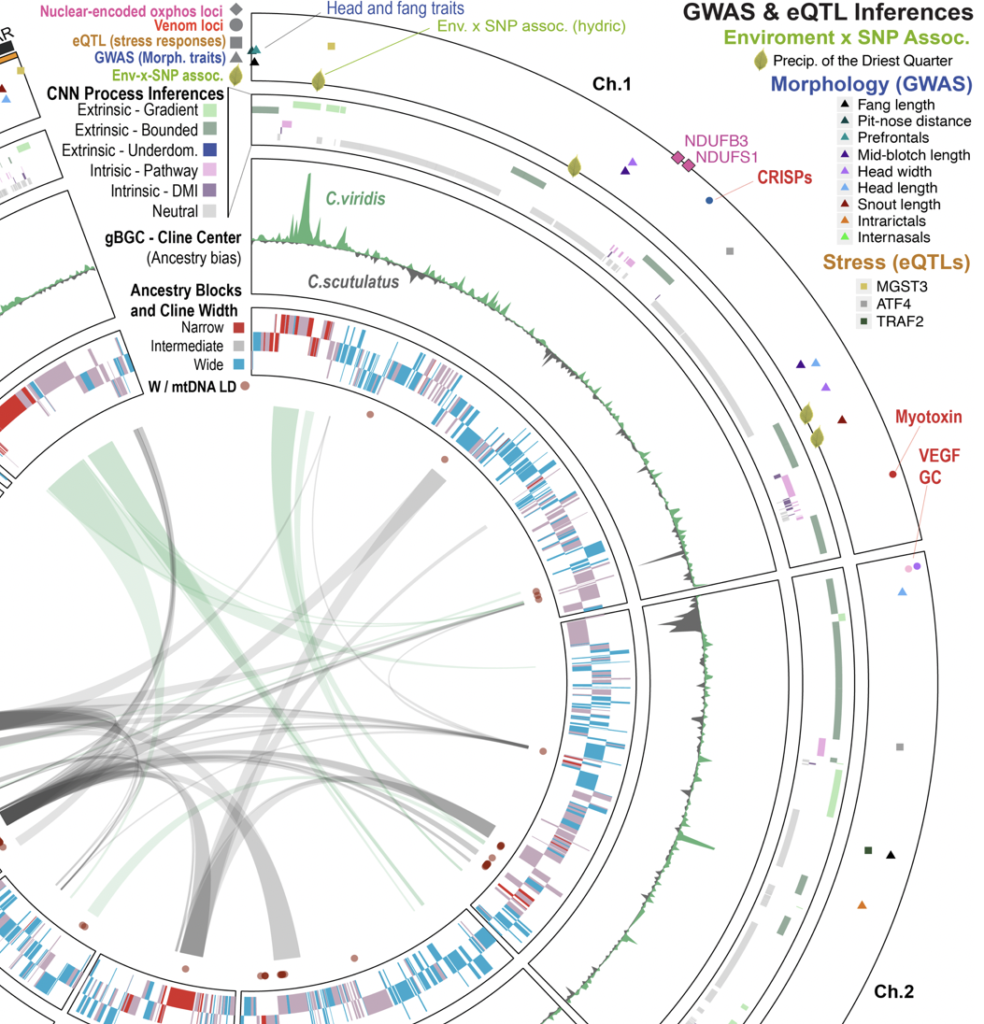
Genome-scale perspectives on the speciation process through comparative analysis of rattlesnake hybrid zones
How do multiple evolutionary processes act synergistically (and antagonistically) to ultimately manifest in reproductive isolation and speciation, and can hybrid zones inform on this process? Hybridization combines divergent genomes into a single organism. Because of this, hybrids face challenges across broad scales of biological organization, from protein interactions at the cellular level, to regulation of divergent developmental and physiological processes, and ecological constraints at the organismal and ecological scale. Evolutionary forces that impact hybrid fitness and hybrid zone dynamics are equally diverse, ranging from ecological selection (gene-environment interactions determine fitness), to negative interactions between genes from divergent parental genomes (gene-gene interactions determine fitness), and novel allelic combinations that increase hybrid fitness. Accordingly, hybrid zones are valuable models for understanding how reproductive isolation evolves through complex interactions of evolutionary processes operating across multiple biological scales, and deciphering these interactions holds great value for understanding the process of speciation and the complexity of genome-trait-environment-fitness relationships. We address these questions using empirical studies conducted on distinct hybrid zones between hybridizing species of rattlesnake species complex that represents a valuable comparative model to other systems with ZW sex determination. This comparative approach facilitates contrasts between lineages at different stages along the speciation continuum, enabling the testing of generalizable “rules” for how multicellular life organizes itself into species. Our work on this project is highly collaborative with a handful of other labs, and includes the development of new machine-learning approaches to link patterns of genomic diversity with evolutionary processes that generate them, and the testing of these inferences using diverse physiological, ecological, and morphological studies.
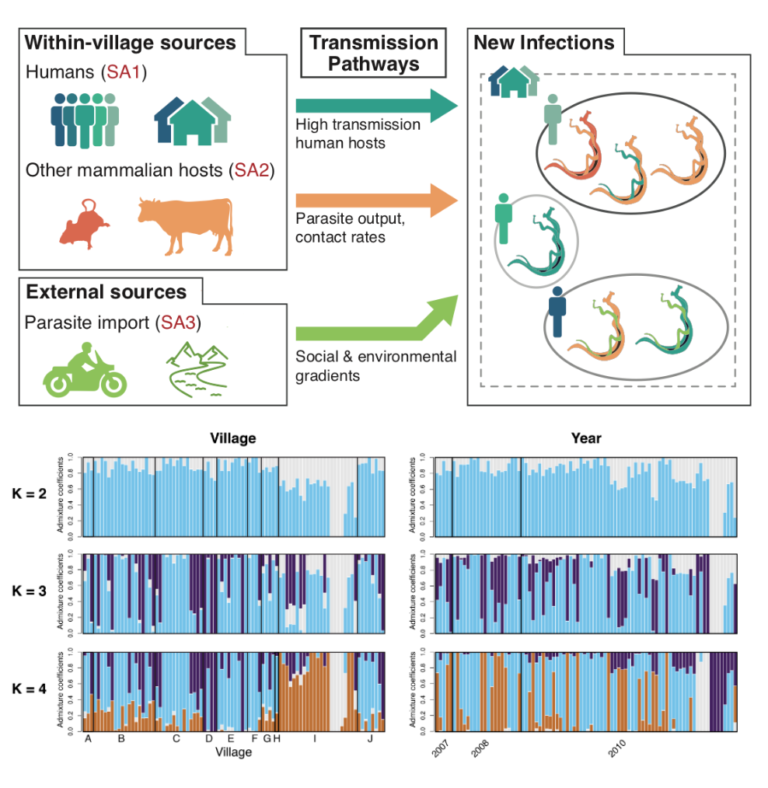
Understanding the roles of transmission and Selection in the persistence of schistosome parasite populations
Why do some populations of parasites persist despite diverse and strong control programs? We are currently applying approaches that what we know well from other projects – analysis of genome-wide SNPs to understanding population genetic structure and genotype-phenotype associations – to understanding the population genetic structure, epidemiology of re-infection, and parasite genotype-patient outcomes for Schistosoma (blood fluke) parasites that cause shistosomiasis in Asia. Working with collaborators studying Schistosoma epidemiology in China, we have developed approaches to collect population genomic-scale data from current and historical shistosome samples to provide retrospective and prospective analyses of outbreak dynamics, basic yet currently not known information on infection biology, and reinfection information. Our goals are to use these data to better understand and thus control shistosome infection, and to use this genomic sampling to detect the evolution of resistance to drugs used to treat shistosomiasis (which would be catastrophic if not adequately prepared for).
EVOLUTIONARY GENOMICS AND LINKING GENOME STRUCTURE, FUNCTION, AND EVOLUTION
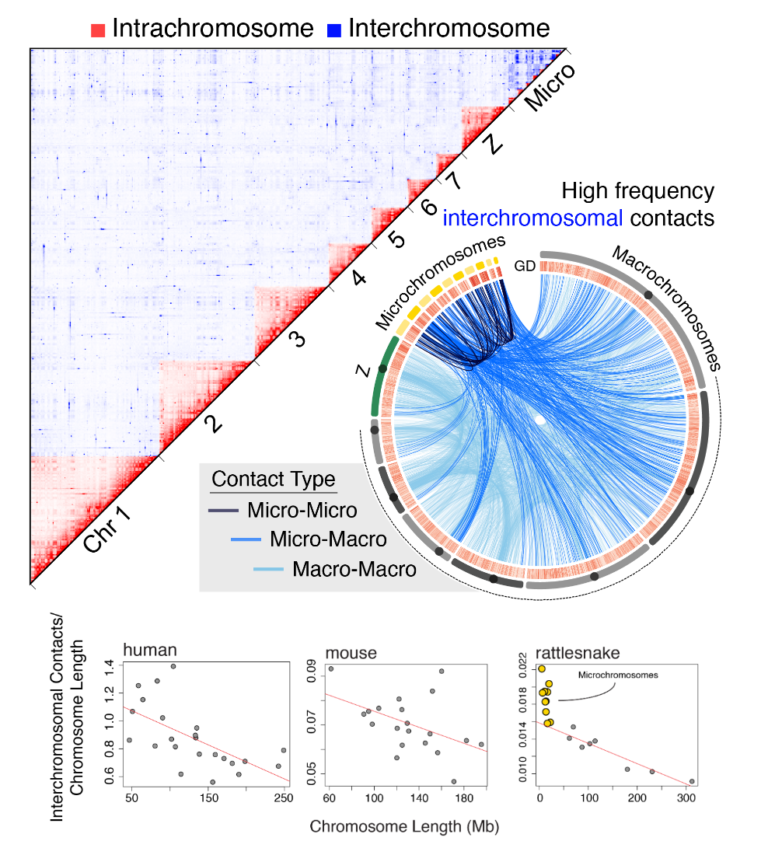
What mechanisms and processes drive the evolution of novelty at the genomic scale, and how do features of genome structure impact genome regulation, recombination, mutation biases, evolution, and function? We are using snake and other reptile genomes to test hypotheses regarding the evolution of complex traits. This work is important for understanding mechanisms underlying adaptation, evolution of complex systems and phenotypes, and the non-canonical function of important vertebrate signaling pathways. Our previous work has provided insight into the unprecedented levels of genome-wide adaptation in snakes, the largest known example of convergent molecular evolution, the evolution and regulation of venom systems, the evolution of vision-related genes in snakes, the evolution of complex toxin resistance, the uniqueness of squamate genome structure and repeat element activity, and the remarkably slow evolution of turtle and crocodilian genomes. Our recent work on snake genomes has exposed a new set of intriguing questions that have motivated new projects in the lab focused on (i) studying the apparently unique features of chromatin structure, function, and evolution of vertebrate microchromosomes and how they drive unique confirmations and functional interactions of chromatin in the nucleus, (ii) the evolution of sex chromosomes in snakes, which we have recently shown is quite dynamic, and (iii) how snake sex chromosomes accomplish regional or gene-specific patterns of dosage compensation that appear to be partially driven by transposable element-derived regulatory elements, and (iv) evolutionary dynamics and consequences of non-canonical PRDM9-driven recombination in snakes.
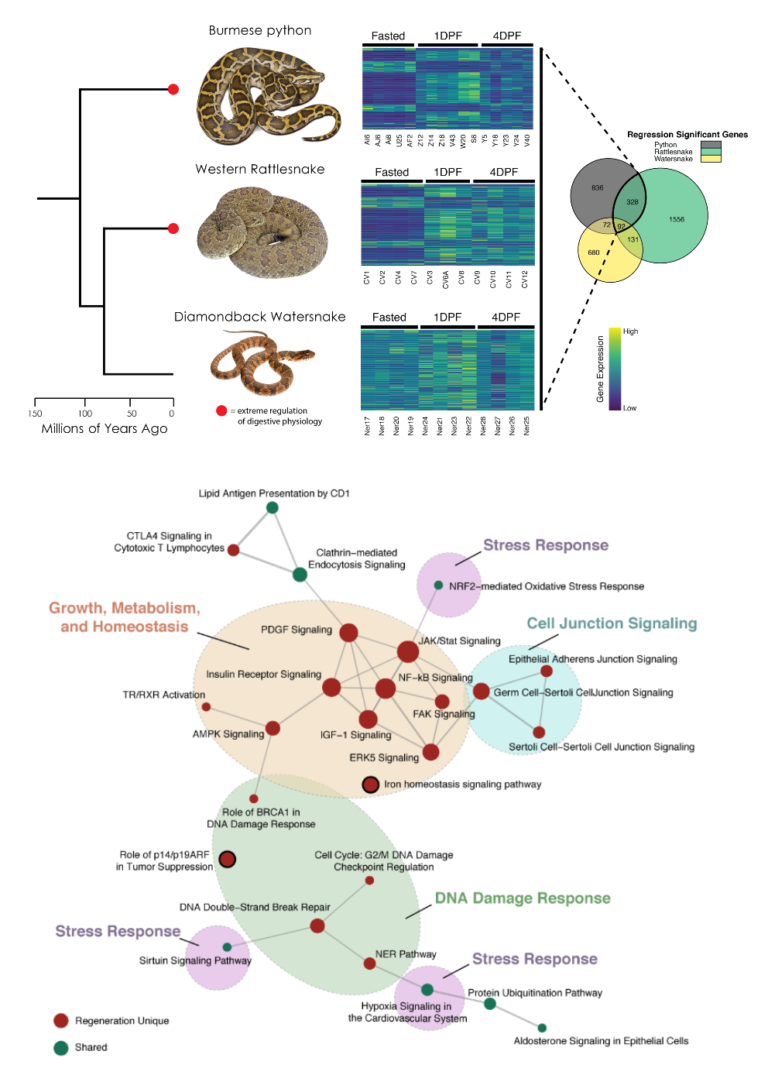
Functional Genomics of Regenerative Organ Growth in Vertebrates
How are conserved signaling pathways in vertebrates evolutionarily co-opted to produce novel phenotypes, and can mechanisms of organ growth in other vertebrates provide insight useful in treating human diseases? Some snakes have evolved the ability to drastically and reversibly remodel their organs, metabolism, and physiology upon feeding. In pythons for example, the heart, intestine, liver and kidneys undergo unprecedented regenerative organ growth (enlarging 40-150% in 48 hours) after fasted animals are fed a large meal. Our work aims to understand the molecular and cellular mechanisms that underlie these reversible growth episodes, and to test if such mechanisms are conserved across vertebrates, including humans – such knowledge would hold great translational potential for directing organ regeneration for treatment of human diseases. One key feature of snakes is that different lineages experience very different regenerative growth phenotypes (and some experience none at all), indicating that snakes hold great comparative potential for dissection of mechanisms that direct regenerative growth. This work has also motivated our sequencing of multiple squamate genomes. Together with several collaborators, we are integrating transcriptomic, proteomic, phospho-proteomic, histological, and single cell data to develop and test hypotheses for the mechanisms underlying regenerative growth responses in snakes, and the degree to which these mechanisms are conserved across vertebrates.